









日前,两款国产新药出海折戟的消息引发了业界广泛关注。
在5.1假期,和黄医药和君实生物先后披露,已收到美国FDA关于旗下创新药上市申请的回复信,对外公告中,美国FDA分别驳回了君实PD-1特瑞普利单抗以及和黄医药索凡替尼的获批申请,前者被要求进行一项质控流程变更,后者被拒的理由则更“严峻”,FDA表示其“已有的临床数据不足以支持获批”。
中国本土药企今后再探“出海”路,应该怎么走?

“这个结果其实并不意外。其实在信达PD-1受挫的时候,单一国家临床数据的问题就引发了讨论。”博济医药子公司美国汉佛莱法规部高级副总裁高翼,具有丰富中美双报经验,他对索凡替尼项目的结果并不意外。

公开资料显示,索凡替尼是一款口服酪氨酸激酶抑制剂,在2019年至2020年间,索凡替尼曾先后获得美国FDA授予孤儿药资格以及快速通道资格。现如今FDA给出的答复称目前基于两项成功的中国Ⅲ期试验以及一项美国桥接研究的数据包,尚不足以支持药品在美国获批。
“种族和地区医疗实践的差异是药物临床结果外推到另一个区域时的重点考量因素,特别是对于美国这种具有非常成熟的创新药审评系统的国家,能否满足本土临床用药需求是其考量新药上市的重中之重。”
高翼表示,国药出海的国际多中心临床研究已是大势所趋。“虽然此次FDA也对君实的CMC部分提出了额外要求,但并未质疑其临床数据。”

然而,这些都是摆在台面上显性功夫,此外还有很多“功夫在诗外”的事情要去做。
“从信达到和记黄埔,我们可以看到FDA越来越关注临床试验入组患者的多样性,临床试验纳入的人群是否具有代表性,以及临床设计能否反映美国的医疗实践这些问题。通常FDA都要求申办方在III期临床之前递交临床多样性计划(Clinical Trial Diversity Plan),这就要求申报方在更早期进行MRCT的布局,并且更早更密切地和FDA保持有效的沟通。”高翼如是说。
事实上,我国创新药不乏出海成功的样本。2019 年11月,百济神州研发的首个国产 BTK 抑制剂泽布替尼获美国 FDA 批准上市,实现中国本土原研抗癌新药出海“零的突破”,这一消息一度让医药圈为之沸腾。两年后,2022年2月,传奇生物宣布其原创CAR-T产品在美获批的消息再度刷屏医药圈,消息公布后,传奇生物在纳斯达克的股价大涨超过12%。

公开资料显示,泽布替尼的首次获批基于两项临床试验的有效性数据,分别为中国开展的单臂Ⅱ期临床研究BGB-3111-206,以及在澳大利亚开展的全球Ⅰ/Ⅱ期临床试验BGB-3111-AU-003,值得注意的是,BGB-3111-206队列中复发难治套细胞淋巴瘤患者的完全缓解率数值达到59%,高于当时已上市的BTK抑制剂伊布替尼或阿卡替尼。支持传奇生物的CAR-T产品最终获批的则是一项1b/II期国际多中心试验,经临床多重治疗后的复发及难治性多发性骨髓瘤患者接受了CAR-T输注,最终结果显示,疾病总缓解率、完全缓解率分别为98%、83%。
然而,这种看似成功的前车之鉴,似乎并不被业界看作是他山之石。因为泽布替尼所使用的单臂临床研究方案,在上月FDA审评PI3K抑制剂中遭到了否定。
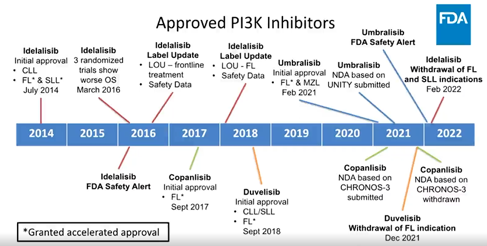
对此高翼也谈及了自己的看法。“FDA批准一款药物是基于对风险和收益的综合评判。RCT从来都是药物有效性的金标准,单臂临床和利用替代终点的加速批准,本身结果就带有很高的不确定性,最终还是要通过IV期临床验证这款药物能否给患者带来生存获益。从这里也可以看出Target Product Profile (TPP)的重要性,一款药物从一开始的差异化定位决定了后续的开发策略,真正解决临床需求的药物才能获得加速开发和批准的机会。”
高翼认为,从单臂临床试验的焦虑其实指向了一个国药出海必须要正视的问题:是否具备产品综合价值差异化的创新能力。
“这个能力验证对于国药出海的临床试验设计和最终结果要求很高,在已有同类产品竞争的情况下,该药必须证明在有效性、安全性以及病人获益率上具有明显优势,对照组试验应首选当前美国的疾病标准疗法,有必要时应开展头对头试验。”
根据相关数据统计,目前至少有13款国产新药向美国FDA提交了上市申请。在接下来的出海之旅中,百济神州及康方生物旗下的两款PD-1产品备受关注,两款PD-1上市申请已获受理,且根据百济神州公告,其PD-1产品上市申请结果预计会在今年7月11日公布。

排着队拿着“出海”号码牌的国产创新药企们会迎来怎样的结局?未来的国药出海如何避免铩羽而归?对此高翼给出了自己的观点:

本文作者系博济医药宣传推广部部长白洋
博济医药“一站式”服务包括:新药立项研究和活性筛选、药学研究(原料、制剂)、药物评价(药效学、毒理学)、小分子创新药一体化服务、临床研究、中美双报(注册服务)、CDMO生产(MAH落地)、技术成果转化等,涵盖了新药研发各个阶段。














