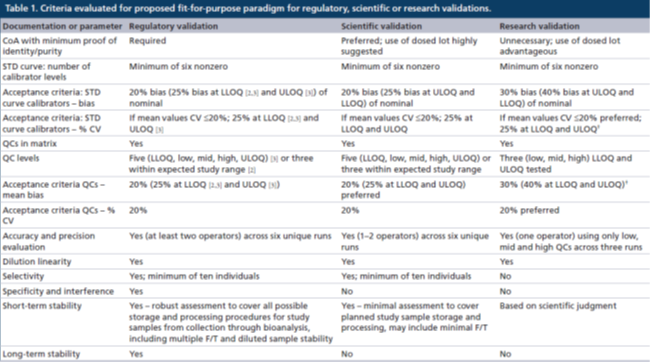
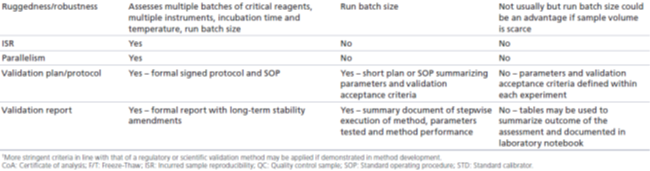
表2. 对于经监管级、科学级或研究级验证的分析方法在研究中样本分析的接受标准

参照(比)标准物
研究级:对于研究级的方法验证,允许使用没有CoA的参照(比)标准物,即比较标准物(comparator),或未表征的标准物(uncharacterized standard),特别是在无法获得完全表征的标准物的时候,例如在药物发现或开发早期。在更早期阶段,由于药物生产和随后的研究的时间安排以及可用的药物数量往往无法对生物药物原液进行完整的表征,为了使人们对分析结果有更大的信心,在可能的情况下最好使用与动物研究中使用的相同批次药物作为参照(比)标准进行样本分析。
监管级验证:对于校准曲线的准确度和精密度的评估应该在6次独立运行中进行且持续几天,在验证期间由多位分析人员执行。
经监管验证的方法必须具有至少6个非零校准点浓度的STD,其中至少75%的校准点浓度的准确度(偏差bias)和精密度(%CV)均应≤20%。在LLOQ和ULOQ校准点水平上:准确度≤25%偏差,精密度≤25%CV。锚定点可以用于取得最佳的曲线拟合,但是其浓度超出了定量范围(ROQ),因此不用于对校准曲线的评估。用于研究前方法验证的非线性曲线拟合和加权方式也必须应用于研究中样本分析,样本分析对STD准确度和精密度的要求与验证阶段相同。不能对LLOQ以下或ULOQ以上的未知样本浓度结果进行外推计算。
监管级验证还要求使用5个质量控制样品(高质量控制点[HQC]、中质量控制点[MQC]、低质量控制点[LQC](在LLOQ校准点浓度的3倍以内)、该方法的LLOQ[LLOQ QC]和ULOQ[ULOQ QC]),或者使用预期研究样本浓度范围内的至少3个质量控制点浓度来评估分析方法的准确度和精密度。偏差和%CV的接受标准均为≤20%,在LLOQ和ULOQ校准品水平上,偏差≤25%,%CV≤25%。QC的可接受总误差(总偏差和%CV之和)为≤30%,HQC、MQC和LQC QC的可接受总误差≤30%,LLOQ QC 和ULOQ QC可接受总误差为≤40%。
在研究中样本分析时,一般使用3个水平的QCs(HQC、MQC和LQC)进行复孔分析,准确度和精密度的偏差和%CV均需≤20%。如果每个校准点(复孔分析)的50%和6个QCs中的至少4个满足上述标准,则该分析运行可以接受,这符合“4-6-20”规则。
科学级验证:在进行研究前科学级验证的过程中,STD曲线评估以及准确度和精密度评估可以由一名或两名分析师在2天内完成,可用来减少研究前阶段的持续时间。这与监管级验证所需要的更稳健评估相反,但是是可以接受的,因为它反映了该方法只是短期地应用于样本分析。科学级验证的研究前验证和研究中样本分析的目标接受标准与监管级验证的目标接受标准一致,但基于数据的预期用途等科学原因,可能会应用更宽泛的接受标准(例如>20%偏差和 %CV >20%)。在后续的"讨论"章节中,所有的接受标准都表示目标/预期接受标准。
在经过科学验证的方法中,校准曲线也应该至少包括6个非零校准点浓度。准确度和精密度的偏差和%CV目标均应≤20%(对LLOQ和ULOQ校准点:偏差≤25%,%CV≤25%)。研究前科学级验证应使用5个级别的QCs (HQC、MQC、LQC、LLOQ QC和ULOQ QC)或者预期研究样本浓度范围内至少有三个QC浓度,准确度和精密度的目标偏差和%CV均≤20%(LLOQ和ULOQ:偏差≤25%,CV≤25%),HQC、MC和LQC的目标总误差为≤30%(ULOQ QC和LLOQ QC为≤40%)。
在样本分析时,应使用三个级别的QCs(HQC、MQC和LQC,复孔分析),其准确度和精密度的目标偏差和%CV均≤20%,如果每个校准点(复孔分析)的50%和6个QCs中的至少4个满足上述接受标准,则该分析运行可以接受,这符合“4-6-20”规则,也适用于监管级验证的分析方法。
应该注意的是,如果在科学级验证的研究前验证阶段,更宽泛的接受标准被证明是合理的,并适用于STDs和QCs的准确性和精密度,那么在样本分析阶段,这些更宽泛的接受标准也应该适用于分析运行的接受。
研究级验证:研究级验证的准确性和精密度以及STD评估应由一个分析人员在3次独立的运行中进行评估,这表示研究级方法验证的严谨性降低了,但这却是可以接受的,因为它反映了该方法只是有限地用于样本分析,以支持内部的早期决策。
研究级验证的研究前验证和研究中样本分析的目标接受标准应该比基于科学级或监管级验证(例如偏差>20%和%CV>20%)更为宽松。更宽泛的准确性和精密度接受标准可用于对STDs的评估(例如30%偏差,在LLOQ和ULOQ可以有40%偏差)。在另一方面,如果STD的表现说明可以达到更严格的接受标准,则应该使用监管级或科学级验证中采用的目标接受标准或者其他更严格但合理的接受标准。
研究级验证中,应评估至少3个级别的QCs(HQC、MQC和LQC),其准确度和精密度的目标接受标准为偏差30%和CV20%以内。这是可以接受的,因为这三个QC的使用提供了对经研究级验证的方法效能的充分控制。如果开发阶段数据表明可以满足更严格地接受标准,则应采用监管或科学级验证标准或其他更严格的接受标准,应该测试LLOQ和ULOQ QCs以供参考,但不需要设立接受标准。
对于使用经研究级验证方法进行的样本分析,如果在研究前验证时,使用了更宽泛的接受标准,则可以扩展用于经监管级和科学级验证的“4-6-20”规则。应该评估3个级别的QCs(HQC、MQC和LQC,复孔分析)。HQC、MQC和LQC的6个稀释QCs中必须有4个回算到其标称值的30%以内(“4-6-30”规则);在接受了的QCs中,必须有4个QCs的%CV≤20%。如果在研究前验证阶段使用了更严格的接受标准,则在样本分析期间也必须采用那些标准。
监管级、科学级或研究级:通常,许多LBA方法的ROQ有限,因此有必要对高浓度样本进行(多次)稀释,以使其进入到ROQ之内。对于所有级别的验证,都有必要证明浓度接近或超过最高剂量水平的预计最大浓度(Cmax),或大于分析方法的既定ULOQ的样本,可以稀释进入既定的ROQ。
为了评估稀释线性度,将已知的待测物加入到空白混合样本基质中来制备浓度在Cmax或附近(或高于ULOQ)的样品,然后稀释这些“稀释线性样品”,使稀释后的浓度落在ROQ之内。如果回算的稀释前浓度在标称浓度的20%以内,则该方法的稀释线性度对于需要进行监管验证的方法是可以接受的。对于科学级和研究级验证,可以使用更宽泛的接受标准 (即验收标准与QC样品相同)。
此外,稀释线性实验还能够识别可能的"钩状效应",需要评估钩状效应(prozone effect)以便为研究中样本分析确立一个合理的稀释方案。在经科学级或研究级验证的方法支持的研究中,分析方法规定的最低稀释倍数(MRD)下如果预期的Cmax不高于ULOQ,那么所有样本分析均可在单一稀释倍数下进行,这意味着可能不需要评估稀释线性度,同一微孔板上QCs的稀释足以证明这一点。
监管级:对于需要监管级验证的方法,在研究前评估生物基质中待测物的稳定性是至关重要的。稳定性评估应涵盖在研究期间所有预期的样本储存条件,包括临床或动物实验期间、运输期间任何中间地点和PK生物分析实验室。长期稳定性必须至少涵盖从样本采集到生物分析完成之前,任何研究样本在相关储存条件下的储存时间。此外,还必须评估预期的样本处理过程中的稳定性,包括冷冻-解冻稳定性、工作台稳定性(或样本处理条件)以及如果在分析前进行样本处理,处理后样本的稳定性。已建立的稳定性应达到或超过研究样本的实际处理时间和条件。至少应该在HQC和LQC水平上评估样本的稳定性,并与新制备的STD和QCs进行比较。如果稳定性样本的分析数值的偏差在15%或20%范围内,则满足稳定性的接受要求。
科学级:经科学级验证的分析方法的生命周期通常很短,而且参照(比)标准物本身的稳定性往往没有很好地建立起来。参照(比)标准物稳定性数据的缺乏也意味着没有必要研究待测物在生物基质中的长期稳定性,建议只进行短期稳定性研究,以便于在样本分析时合适地存储和处理研究样本。因此,在研究前验证阶段,通常建议在计划的储存条件下进行有限的冷冻-解冻测试和短期的稳定性研究(数周或数月,而不是数年)。如果在研究开始前,预知需要某些独特的存储环境,则应在研究前阶段评估那些条件下的稳定性。与监管级验证一样,至少应在HQC和LQC水平上评估稳定性在科学级验证中使用的接受标准,可用于稳定性样品(但是如果对验证QCs使用更宽泛的接受标准,那这也适用于稳定性样品)。
研究级:经研究级验证的方法的生命周期特别短,因为许多使用这些方法的研究也是短期的。参照(比)标准物的稳定性一般未知,因此也不需要长期稳定性数据,常规地(routinely)评估短期稳定性,应该只有在对待测物的经验有限的情况下进行。如果存在已知或疑似的药物不稳定性,那么了解它们对样本分析结果的影响很可能是有益的。因此,建立样本的基本稳定性如冻融或有限的工作台稳定性,以进行样本处理,会有助于获得可靠的分析结果。如果需要评估稳定性,至少应该在HQC和LQC水平上进行,研究级验证中使用的接受标准应适用于接受稳定性样品(如果对验证QCs使用更窄的接受标准,这也适用于稳定性样本)。
监管级:相关指南文件没有直接探讨稳健性和耐用性评估。但是,从行业的最佳实践白皮书中可以得知,监管级验证经常有包括此类评估,以确保该方法在实验室通常可能发生的条件下产生可重现的结果。评估稳健性时,需要引入特定的变化(如孵育温度、孵育时间、光照或基质),然后研究该方法的一致性(consistency)。此外,一般通过研究下述内容来评估该分析方法的耐用性:正常进行分析过程中可能出现的不同操作条件,如不同的分析人员、不同的仪器或试剂等,建议进行运行大小的测试(batch size testing)。稳健性和耐用性评估应该反映预期的操作条件和潜在的变化,而该分析方法在其整个生命周期内会在这些操作条件下或潜在变化真的发生时被使用。
科学级:经科学级验证的方法,不需要像监管级验证方法那样在多个分析人员、设备和持续时间上展示相同水平的稳健性,因为这些分析方法的生命周期更短,因而预期的变化更少。经科学级验证的方法在进行样本分析的实操条件时,稳健性和耐用性评估会帮助提供足够的信心来使用该方法,但无需在这些条件之外进行更多的评估。因此,如果一个分析人员正在对有限的设备进行研究前验证和研究中样本分析,并有足够的试剂来完成整个分析任务,则稳健性评估可能仅限于运行大小的测试,以确保微孔板的批量处理足以支持样本分析。
研究级:由于经研究级验证的方法的应用非常有限,因而对研究级验证的分析方法一般不需要评估其稳健性和耐用性,但如果要分析大量的或体积有限的样本,则评估运行大小(batch size assessment)可能是有益的。
监管级:必须证明需要监管级验证的分析方法对待测物具有选择性,以证明基质成分不会干扰分析结果。应该使用至少10个单体的空白生物基质,并在LLOQ水平上外加该生物药,以评估分析方法的选择性。评估外加待测物(接近LLOQ浓度)样品的回收率,以确保基质效应不影响该方法在上述浓度的回收率。如果可能,建议在相关疾病状态的生物基质中评估回收率,并且也对基于血清或血浆的分析方法、对高血脂或溶血样本评估回收率。如果在10个样本中8个外加待测物的回收率在标称值的25%的偏差范围内,并且对未外加待测物的样本所测定的待测物浓度小于LLOQ,则该分析方法的选择性评估符合接受标准。如果此评估的结果,在方法开发阶段发现不可接受,则建议在验证之前提高LLOQ的浓度。
科学级:需要经科学级验证的方法应该评估其选择性,与监管级验证一样,评估应包含10个单体的生物基质样本,并且要评估外加和未外加待测物的样品。相反,并不需要对高脂质或溶血样本(血清或血浆样本)评估选择性。另外,只有在疾病状态已知会引入干扰物质的情况下,才建议评估疾病状态的生物基质。接受标准与监管级验证一致:80%的样本的回收率偏差需要在25%之内。
研究级:对于研究级验证,不需要全面地评估选择性,因为在测试LLOQ QC样品时,已经在混合基质中进行了近似的选择性评估。如果某个动物中存在所服用的药物的内源性同源物或其他有科学原因,则可在较少数量的单体基质样本中评估选择性。用于STD曲线中LLOQ的接受标准也适用于选择性样品。
监管级:对于需要监管级验证的方法,需要证明用于测试的试剂仅与该生物药即待测物结合,并且不与其他相关化合物或伴随药物发生交叉反应。应在该方法的LLOQ和ULOQ水平上,用QCs评估特异性。例如结构相似的生物药、抗药物抗体、可溶性药物靶点、已知的伴随药物等,偏差在25%以内的QCs即满足可以接受的特异性。应将已证明的干扰物质记录在案。另外,如果干扰物妨碍了PK数据的适当解释,则需要在方法验证之前,采取措施(如重新开发该方法)以减轻其影响。
科学级:对监管级验证的方法,必须评估其特异性,但对于科学级验证来说则不是必须的。一般来说,需要科学验证的方法无需费力地开发分析方法,以减轻来自抗药物抗体(ADA)或可溶性靶点的干扰,因为严格证明该方法可接受的要求太高,除非相关干扰非常普遍,且妨碍了基本的PK数据解释。
研究级:可省略特异性测试。对研究级验证的方法,无需全面地描述或减轻干扰,因此不需要评估方法的特异性。
监管级:由于研究前验证所用的STDs和QCs可能不能完全模拟研究样本的性能,作为监管机构的要求,ISR是为了确保所报告的样本浓度的可靠性,且视为准确性的模拟评估。对于1000个样本以内的研究,ISR需要测试10%的样本;而对于1000个样本以上的研究,应进行5%的ISR测试或者测试7%的所有研究样本。这些样本应来自Cmax附近和药物消除阶段。在这一评估中,ISR样本在另一个日期重新测试(重复repeat),以模拟最初的生物分析(原始original)。该评估比较重复(repeat)和原始(original)测定结果之间的百分比差异,如果二者结果相差≤30% ,则认为样本的测定结果是可以接受的。如果2/3的测试样本(67%)符合样品接受标准,则整个评估合格。如果评估失败,必须进行调查以确定失败原因,并采取适当措施,尽量减少不准确的结果。
此外,还应该使用ISR样本评估方法的平行性。平行性在概念上类似于稀释线性,但使用实际研究样本进行连续稀释,单个样本之间的个别基质成分的差异可能会干扰测定结果。虽然FDA指南草案规定,“稀释后研究样本的平行性应使用稀释后的校准品进行评估,以检测基质效应”,但没有就接受标准提出指导。EMA指南对评估样本平行性的期望提供了更多信息:"应使用空白基质稀释高浓度的研究样本(最好接近Cmax)到至少三个浓度。稀释系列中样本之间的精密度不应超过30%。”因此,应选择在Cmax附近的研究样本并连续稀释,使得至少三个稀释浓度在方法的ROQ内。如果稀释结果回算后浓度的%CV≤30%,则样品符合验收要求。如果评估失败,必须制定一个计划,说明如何报告稀释样本的测定浓度。
科学级:对于经科学级验证的方法,常规的话不会在研究中对ISR和平行性做一步的验证/评估。如果在研究中样本分析过程观察到样本稀释的非平行性,则可以按照与监管级验证相同的设计和接受标准来评估平行性。监管级验证的指南文件规定:对支持人体生物等效性研究、关键PK或PD研究以及非临床安全性研究必须评估ISR和平行性,这些研究的结果将被纳入向监管机构提交的文件。科学级验证并非是为了长期应用该分析方法而验证的,使用该类方法的主要目的是支持非GLP(non-GLP)或非关键性研究,这些研究的数据不会用于支持监管申报文件中的主要结论。
研究级:经研究级验证的方法仅会短期使用,主要用于候选药物的选择或早期临床前研究,其数据仅用于内部决策。因此,不需要评估ISR和平行性,除非特别有必要。
本文如有疏漏和误读相关指南和数据的地方,请读者评论和指正。所有引用的原始信息和资料均来自已经发表学术期刊, 官方网络报道等公开渠道, 不涉及任何保密信息。参考文献的选择考虑到多样化但也不可能完备,欢迎读者提供有价值的文献及其评估。